Blu Genes Foundation Impact - Research and News, NEWS UPDATE
UMass Med School gene therapy shows promising early results in tackling Tay-Sachs

UMass Medical School Dean Dr. Terence R. Flotte is interviewed in his office about a UMass study on investigational genet therapy in two patients with infantile Tay-Sachs disease. [Photo/Sam Fuller]
WORCESTER — The fight against Tay-Sachs disease, a rare, progressive and fatal neurodegenerative disorder, showed progress based on preliminary results from a University of Massachusetts Medical School expanded access study presented last month.
This was the first time that Tay-Sachs gene therapy has been done in humans, as opposed to animal studies, and the first time gene therapy to correct the enzyme deficiency that causes Tay-Sachs has been inserted safely into the brain, according to Dr. Terence R. Flotte, dean of the medical school and Celia and Isaac Haidak professor of medical education.
“Those were the two big things,” he said in an interview.
The next step will be to test the therapy in increasing doses on more patients. An investigational new drug proposal has been submitted to the U.S. Food and Drug Administration and a phase 2 clinical trial is expected to start within a few months. Flotte will be the principal investigator in that study.
Axovant Gene Therapies, a Swiss company developing gene therapies for serious neurological diseases, last year licensed exclusive worldwide rights for the development and commercialization of the novel gene therapy programs.
Flotte reported at the European Society of Gene and Cell Therapy Annual Congress in Barcelona, Spain, that two patients with infantile Tay-Sachs disease, who were treated at UMass Memorial Medical Center with gene therapy developed at the medical school, showed signs that progression of their disease was modified.

Mona Vogel of Groton and her 6-year-old son Owen, who has juvenile Tay-Sachs. [Submitted Photo]
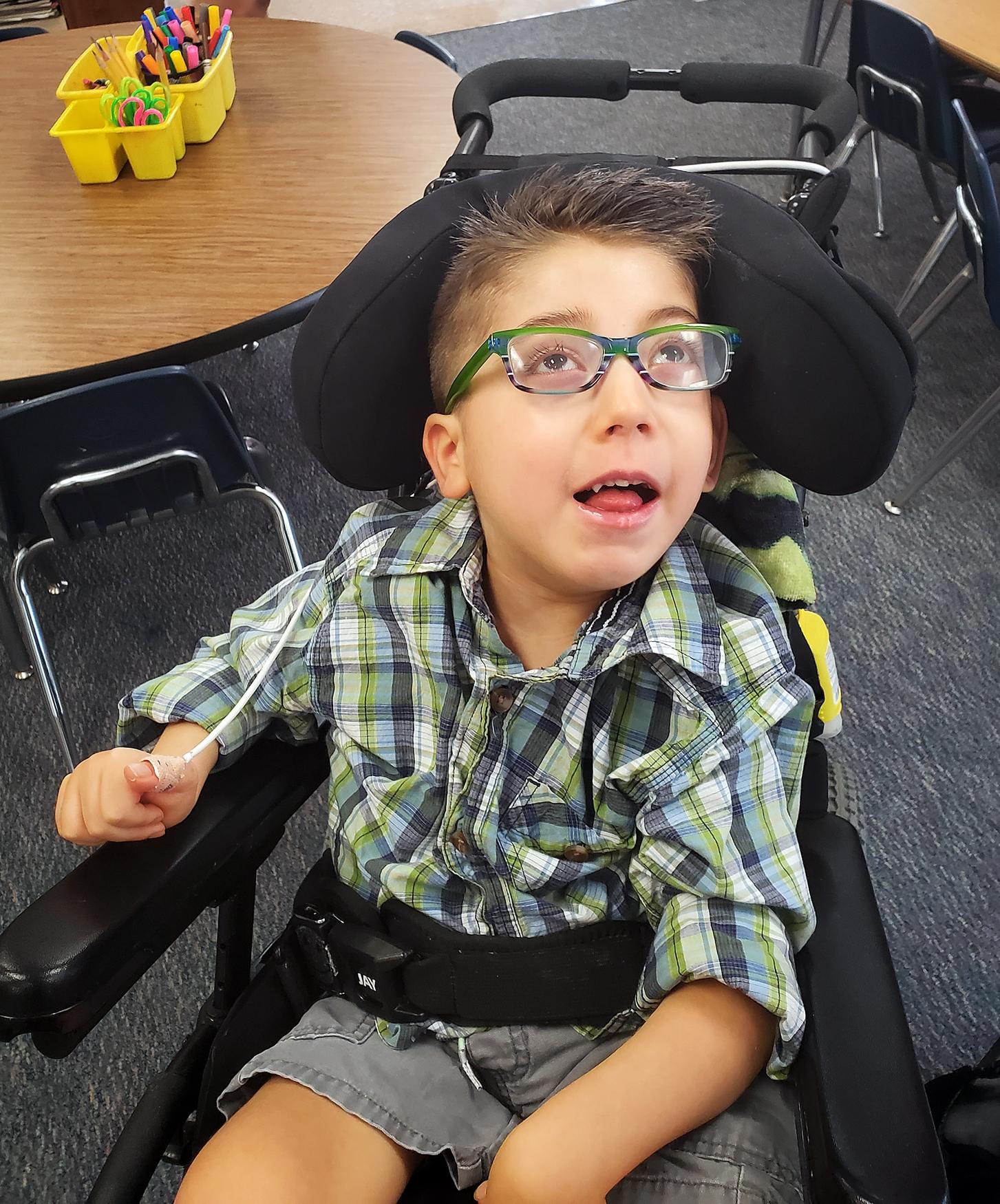
Owen Vogel, 6, has juvenile Tay-Sachs. [Submitted Photo]
The first patient, who has advanced disease, received treatment a year ago, at around age 2½. An engineered virus containing corrective genetic material was injected into fluid surrounding the brain. The patient hasn’t shown clinical improvement in functioning but biochemical changes were detected in the brain, indicating partial re-creation of the missing enzyme associated with Tay-Sachs, Flotte said.
The second patient, who was around 6 months old when some of the gene therapy was injected into the thalamus region of the brain, about six months ago, has not degenerated further since then.
Flotte said the thalamus is “the master relay station for the brain,” which allows the genetic material to spread to other parts.
Tay-Sachs, which results from the absence of beta-hexosaminidase (HexA) enzyme, is diagnosed in about 30 children in the United States each year, and there are an estimated 400 to 700 cases worldwide, according to medical school reports.
According to the Cure Tay-Sachs Foundation, the Tay-Sachs gene is carried by one in 27 Ashkenazi (Eastern European) Jews, French-Canadians or Louisiana Cajuns; one in 50 Irish-Americans; and one in 250 in the general population. If both parents carry the Tay-Sachs gene, there is a 25 percent chance their child will suffer from Tay-Sachs and likely die at a young age.
There is no cure for the disease.
In infantile Tay-Sachs, Flotte explained, babies start to develop typically, reaching milestones such as sitting up. But then as the disease progresses, they lose that ability and face other developmental challenges.
“A home run would be to maintain the ability to sit and gain the ability to walk,” Flotte said. “Helping babies to gain those developmental milestones is really the goal.”
Both patients in the study showed indication of enzyme being made in the brain after introduction of the therapy. But Flotte said it was too soon to know if stabilization of their condition will prolong life expectancy.
The median life expectancy for children with infantile Tay-Sachs or a similar inherited neurological disorder, Sandhoff disease, is about three to five years.
The process to insert the gene therapy with millimetric target precision into the skulls of very young children required extensive planning and computer modeling ahead of time, pediatric neurosurgeon Dr. Oguz Cataltepe said. A safe trajectory had to be mapped so that blood vessels wouldn’t be harmed. A robotic arm was used with the insertion.
Patients also had to be given drugs to suppress their immune system so they wouldn’t reject the gene therapy.
Cataltepe said the first patient’s procedure took about two hours. Subsequent insertions would take longer as more of the therapy is injected directly into the brain.
Flotte said the families faced the daunting procedure with “trepidation, but I think some level of hope.”
He said families felt because of the outcomes of Tay-Sachs in its natural rapid progression, it warranted the risk.
“I think both of them have been very grateful to try the technology available,” said Flotte. Still, “They recognize these are very early steps.”
He added that neither family in the study knew of any hereditary risk. Neither has Ashkenazi Jewish heritage, a group among those with the highest risk.
Flotte said he would like to see Tay-Sachs disease be part of standard newborn screening programs, particularly if therapy becomes available.
Mona Vogel of Groton, whose son, Owen, 6, was diagnosed with juvenile Tay-Sachs when he was about 3, also urges people to get screened for the disease.
Vogel, a single mother by choice, is not of Ashkenazi Jewish descent. She went to two different genetic counselors, and her risk for Tay-Sachs didn’t come up. Her donor’s genetic profile also didn’t highlight a risk.
Owen developed typically until he was 3, Vogel said in a phone interview. “Then he started falling — face-plant falling.”
Vogel is active within the Tay-Sachs community and said there had been “tentative excitement” over apparent progress in previous animal studies, often to be met with disappointment in setbacks.
“There’s this combination of excitement and reserved excitement,” she said about news that a human gene therapy study shows promise.
The scary part for parents of children with Tay-Sachs is that they don’t yet know what the criteria will be for inclusion in the clinical trial and whether they will get a chance to participate.
“To come this far and not even have this opportunity is devastating,” Vogel said. “There’s a lot of consistently heightened emotions that’s a mixed bag of all of the above.”
Children accepted into the clinical trial will be treated with gene therapy at UMass Memorial, Flotte said. Their progress will be evaluated at Massachusetts General Hospital in Boston, for independent external assessment.
Vogel said she’s trying to remain positive.
“For me, I would be perfectly willing to risk it all (to be part of the research),” she said. “Just so I know we did everything we could do. And I promised this kid I’d do it.”
“As the mother and aunt of a daughter and nephew who both died from infantile Tay-Sachs nearly 20 years ago, I am so grateful to the team at UMass that advanced the research towards treatments and where we are today,” wrote Blyth Lord of Newton, founder of Courageous Parents Network, in an email.
“In addition to the team at UMass, I credit families and the patient disease groups National Tay-Sachs and Allied Disease (NTSAD) and Cure Tay-Sachs that have channeled family support to research. You need the families, the patient disease groups, the researchers and the money to make this possible. Of course, we know this first phase therapy is early stage and there is still a long way to go, so we will all have to keep going too.”
Researchers who collaborated on animal models and therapeutic approaches for Tay-Sachs and similar disorders also include: Miguel Sena-Esteves, associate professor of neurology at UMass; Dr. Heather Gray-Edwards, formerly of Auburn University and currently assistant professor of radiology at UMass; and Douglas Martin, professor of anatomy, physiology and pharmacology in the College of Veterinary Medicine and the Scott-Ritchey Research Center at Auburn University.
READ THE FULL ARTICLE HERE
![]()